Winter 2026: Keely Gibb defended her PhD thesis with a nomination for a senate medal! Sean Kirkby defended his MASc thesis on "Applying Deep Learning for Monitoring Mobility Aid Usage from LiDAR Data". Anthony Fuller's paper on "Thicker and Quicker: A Jumbo Token
For Fast Plain Vision Transformers" has been accepted to ICLR2026.
Fall 2025: Oly Papillon defended his MASc thesis with a nomination for a thesis medal. Mohsen Mozafari defended his PhD on "Remote Respiratory Monitoring from Video Data". Francois Charih defended his PhD and was nominated for a Senate Medal. Congratulations to all!
Summer 2025: The lab enjoyed a beautiful day at the cottage this summer. Kade received an OGS scholarship for 2025-26! Francois has defended his PhD entitled "Sequence-based Peptide Binder Design to Modulate the Lysine Methylome and Beyond" (co-supervisor: Prof. Kyle Biggar). Francois was nominated for a Senate Medal for the excellence of his thesis. Mohsen Mozafari has also defended his PhD on the topic of "Remote Respiratory Monitoring from Video Data". Anthony and Yousef have co-led a NeurIPS2025 paper on "LookWhere? Efficient Visual Recognition by Learning Where to Look and What to See from Self-Supervision"
Winter 2025: Coming soon!
Fall 2024: Anthony Fuller has had his second NeurIPS paper accepted! (co-authored with Daniel Kyrollos and Yousef Yassin)
Summer 2024: Yousef Yassin received silver and gold medals. Writeup: https://carleton.ca/sce/2024/spotlight-on-yousef-yassin-a-rising-star/
Spring 2024: Coming soon!
Winter 2024: Coming soon!
Fall 2023: Anthony Fuller has a paper accepted to NeurIPS 2023, representing the first paper in the main NeurIPS conference for Carleton University (I believe).
Spring 2023: Victoria Ajila successfully defended her MASc thesis entitled "Adapting Genome-wide microRNA Discovery and Target Prediction to Specific Species". Victoria's thesis has been nominated for a Senate Medal. Our research relating to non-contact patient monitoring in the NICU has been recently highlighted after a presentation to parliamentarians. Anthony Fuller has received an NSERC PGS-D scholarship to support his PhD research!
Winter 2023: Jonathan Dupuis has joined our CUBIC lab to conduct research on applications of ML to critical electrical infrastructure monitoring.
Fall 2022: With COVID19 restrictions relaxing, our lab took at break for some indoor rock climbing. Jim Green served as an external examiner for an ML-related application as part of the CIHR Fall 2022 Project Grant competition. Anthony Fuller's paper on "SatViT: Pre-training Transformers for Earth Observation" has been accepted for publication in IEEE Geoscience and Remote Sensing Letters.
Summer 2022: PhD student Kevin Dick has won the Governor General's Gold Medal for his PhD thesis. In partnership with Natural Resources Canada, Carleton University will undertake 10 months of research in the area of "Multi-Sensor Applications To Address Critical Electricity Infrastructure Vulnerabilities". Several students in the lab are working on this research initiative. Jim Green delivered a machine learning workshop as part of the "Digital Exploration Opportunities Program" (DEOP). This program has been recognized with “Electricity Resources Branch Director General’s Award” at Natural Resources Canada. Jim Green is a collaborator on a successful CIHR grant entitled "Artificial Intelligence for the Prevention of Unplanned Dialysis". This project is being led by nephrologist, Dr. Greg Hundemer, and will fund the PhD research of Martin Klamrowski and Elmira Amooei. Francois Charih's paper on "Assessing sequence-based protein-protein interaction predictors for use in therapeutic peptide engineering" has been accepted to Scientific Reports. Kevin Dick led a paper with a team of students published in Scientific Reports entitled "Reciprocal Perspective as a Super Learner Improves Drug-Target Interaction Prediction (MUSDTI)".
Spring 2022: Kevin Dick and Yasmina Souley Dosso have defended their PhD theses! Eric Arezza has defended his MASc thesis! Our lab will be publishing papers at IEEE I2MTC and IEEE MeMeA this spring. Kevin Dick's PhD thesis has been nominated for a Senate Medal. James Green presented a 40-minute workshop on "Machine Learning (ML) Applications in Critical Infrastructure Monitoring" as part of the CEATI Virtual Black Sky Hazards Workshop. Congratulations to incoming MASc student, Anthony Fuller, who has received a Vector AI Scholarship to support his graduate research in Vision Transformers for Remote Sensing. Congratulations to Victoria Ajila who received an OGS scholarship.
Fall 2021: PhD student Yasmina Souley Dosso has had a paper accepted to Elsevier Computers in Biology and Medicine on the topic of "RGB-D Scene Analysis in the NICU". PhD student Kevin Dick has published in the Journal of Proteomics with collaborators at Agriculture Canada. Kevin and Daniel had a paper accepted at SPLASH-E describing the integration of Machine Learning research into an undergraduate course. Our lab will also be getting together for a hike in Gatineau Park in October as we transition to post-COVID19 life.
Summer 2021: PhD student Kevin Dick has won the Outstanding TA Award for SYSC4906 - Intro to Machine Learning. Apparently 274 TAs were nominated and only 5 awards were given, so congratulations Kevin! Congrats also to multiple lab members who have had conference papers accepted this summer (Kevin/Josh/Francois CRV2021, Yasmina EMBC2021, Mohsen/Daniel/Yasmina/Josh SAS2021.
Winter 2021: Congratulations to MASc student, Daniel Kyrollos, for winning 3rd place in the Data Day 7.1 poster competition for his poster on A Meta-Model in NLP for Hatefulness. Daniel will also receive an Ontario Graduate Scholarship next year. Kevin Dick was the lead author of a PeerJ paper on "Multi-schema computational prediction of the comprehensive SARS-CoV-2 vs. human interactome". Postdoc Dr. Roy Wang was lead author on a review paper in Metabolites on "Automatic 1D 1H NMR Metabolite Quantification for Bioreactor Monitoring". Kevin Dick and Daniel Kyrollos both published papers in IEEE I2MTC 2021. Kevin Dick participated on an expert panel at Data Day 7.1 on the topic of "Smart Everything". Fadwa Darwaish and Roger Selzler both had papers accepted to IEEE MeMeA 2021.
Fall 2020: Our NSERC Alliance-funded COVID19 research has been higlighted in a recent story.
Summer 2020: Congratulations to MASc student, Fadwa Darwaish, and the entire IEEE WIE executive team for winning the 2020 WIE Affinity Group of the Year Award!. Congratulations to MASc student, Daniel Kyrollos, for being awarded a Vector AI Scholarship to support his research in neonatal patient monitoring! Congratulations to MASc student, Samreen Aziz, for winning "Best Student Paper" at the 2020 IEEE MeMeA conference! James Green was interviewed on CBC Radio 1 regarding our lab's research to fight COVID19 through peptide research. Together with Profs Robert Langlois, Adrian Chan, and Stephanie Redpath, we have received $522,000 over three years to fund research into reducing vibrations during neonatal patient transport.
CUBIC: Carleton University Biomedical Informatics Co-laboratory
At the Carleton University Biomedical Informatics Co-laboratory (CUBIC), we apply machine learning and data science to solve problems in biomedical informatics. We are particularly interested in predicting rare events, or conducting machine learning in the presence of class imbalance. Current projects requiring additional students include an exploration of the use of RGB-D video and pressure-sensitive mats for real-time patient monitoring in the NICU at CHEO, characterizing and mitgating vibrations experienced by neonatal patients during emergency ground and air transport to the NICU at CHEO, and development of novel machine learning methods for analyzing protein structure, function, interaction, and chemical modification. Interested students should have strong software and communication skills proven through academic performance and/or industry experience. Hands-on experience with software development, mahcine/deep learning, computer vision, web development, and statistics is highly valued.
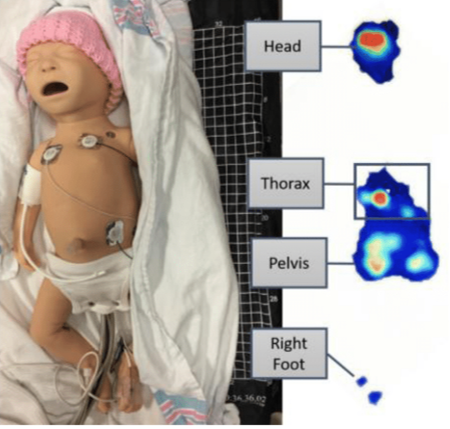
Non-contact Neonatal Patient Monitoring We are working with CHEO to investigate novel patient monitoring technologies in the NICU. We are examining the use of pressure-sensitive mats (PSM) to provide continuous, unobtrusive, and non-contact monitoring of critically ill babies in the neonatal intensive care unit (NICU) at CHEO. We are working with IBM's Centre for Advanced Studies (IBM-CAS), Dr. JoAnn Harrold (Neonatologist @ CHEO), and Mr. Kim Greenwood (Director of Clinical Engineering @ CHEO). In addition to PSM, we are also using multispectral cameras (colour, near-infrared, and depth) to record video of patients from above. From the PSM and video data, we are developing deep learning computer vision approaches for physiological monitoring (HR, RR, etc), characterizing patient movement, and detecting clinical interventions with an eye on semi-automated charting. We have developed a tablet app, such that bedside researchers can annotate all events of interest for up to 6 hours per patient. These gold-standard annotations will be used to develop and validate machine learning approaches to semi-automated non-contact patient monitoring in the NICU.
Estimation of Patient Stress during VR-Therapy Together with Prof. Adrian Chan and clinicians from the Canadian Forces and the Ottawa Hospital Rehabilitation Centre, we are developing novel methods for estimating patients' stress levels (sympathetic activation of the autonomic nervous system) during virtual reality therapy for PTSD, mild traumatic brain injury, and complex pain. Such monitoring will enable clinicians to tailor the intensity of therapy to induce brain plasticity and healing, while avoiding over-stimulating the recovering brain. We are using both gait information, from a VICON motion-capture system, and physiologic signals from a wearable sensor. Deep learning models will be used to analyze these data to generate estimators of stress in real-time, thereby providing an important tool to rehabilitation clinicians.
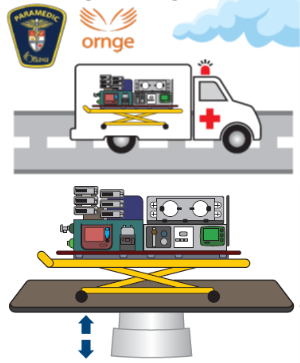
Quantifying Patient Vibrations During Emergency Transport Each year, thousands of newborns in Canada are transported by air or ground ambulance to receive specialized medical care. These infants are often premature and especially vulnerable during transport. Vibration exposure during transport may contribute risk of serious long-term consequences including brain injury. To decease risk during transport, specialized equipment is used. However, vibration levels experienced by infants and contributing factors are not well understood. This lack of knowledge affects the ability to transport newborn patients in the safest manner possible. With Rob Langlois (MAE), Adrian Chan (SCE), and Stephanie Redpath (CHEO), we are seeking to increase the safety of infant transport by reducing vibration exposure. We propose a research program which seeks to understand how vibrations caused by the road and air environments are transferred to the infant. This will allow us to propose novel methods to reduce vibration exposure and improve the equipment for transporting fragile infants. We will measure vibrations in many transport scenarios including within hospitals as well as in ground ambulances, helicopters, and airplanes. The results will be used to develop test equipment and procedures for evaluating the transport equipment. This research will enable better understanding of the problem and provide a reliable way to test the transport equipment and procedures. It will lead to new tools for planning routes that minimize vibration and for monitoring patient vibration.
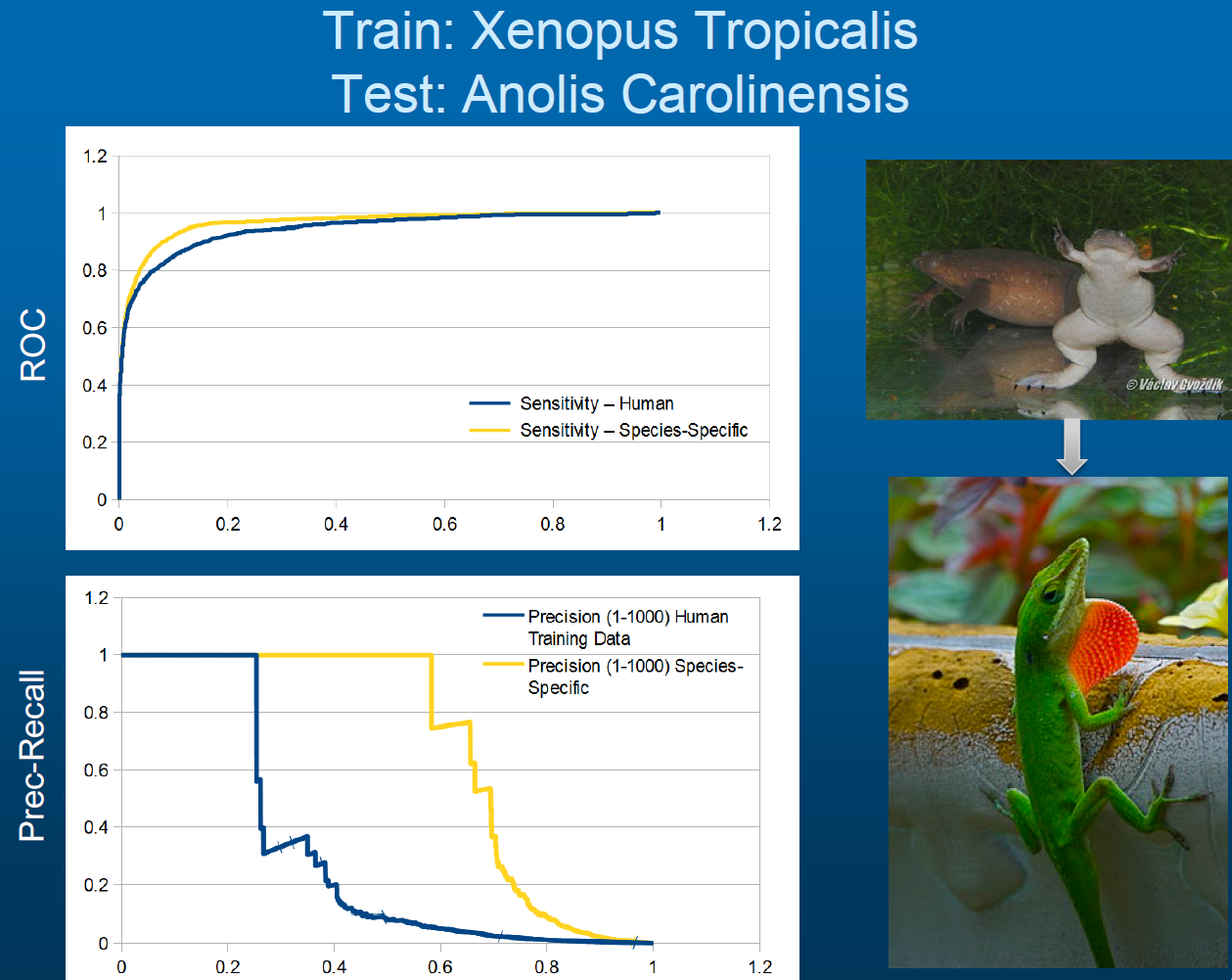
Semi-Supervised and Species-Specific Prediction of microRNA. microRNA are short RNA molecules that play an important role in post-transcriptional gene regulation. Our collaborators are continuously sequencing new species and wish to identify novel microRNA within these new genomes. However, most widely-used microRNA prediction tools are only effective on human data. We have developed SMIRP, a framework for the creation of species-specific predictors of microRNA from genomic sequence. We have achieved up to 500% increases in sensitivity at precisions of up to 90% when compared with existing methods. SMIRP has been applied to study numerous genomes including turtles, slime moulds, and a snail. We are now developing methods to leverage transcriptomic RNA-Seq data as this continues to becomes more accessible to experimental researchers.
Post-translational modification. While progress continues to be made on the prediction of structure from sequence, knowledge of a protein's structure may not be sufficient to discern its function. For example, most proteins undergo some form of post-translational modification (PTM) following initial synthesis which may have a profound impact on protein function. Our lab is therefore working to develop intelligent predictors of important PTM's such as sumoylation and phosphorylation. Iterative prediction of protein function and structure is a long term goal as well.
Check out our YouTube channel for recent conference presentations by members of the cuBIC lab.
Bioinformatics web services. Please click here for a partial list of web services developed by our lab.
Areas of Research Interest
My research focus has been in the following areas:
Real-time patient monitoring, using pressure-sensitive mats, multi-modal video, and other sensors.
Machine learning, pattern classification, data mining
Bioinformatics, proteomics, and prediction of protein structure, function, interaction, and post-translational modification
Development of novel assistive technology and devices
Current projects include:
Neonatal patient monitoring using pressure sensitive mats (collaboration with IBM-CAS)
Classification of audiograms for tele-medicine applications (collaboration with Clearwater Clinical)
Real-time monitoring for vibration/acceleration, sound, air pressure, and temperature during emergency patient transport to the NICU (collaboration with the CHEO patient transport team)
Semi-supervised and species-specific prediction of microRNA from genomic sequence or transcriptomics data
Prediction of protein-protein interactions from sequence
Development of various novel assistive devices for persons who are disabled and the elderly
Identification of post-translational modifications in proteins, including methylation, sumoylation, glycosylation, and hydroxylation.